

Multiple Endocrine Neoplasia Type 2 (MEN2 Syndromes)
MEN2 is a very uncommon syndrome of endocrine tumours that affects about 1 in every 30,000-50,000 people. It is passed on as an autosomal dominant inheritance (see MEN Genetics for an explanation of the genetics of MEN syndromes), and is caused by a mutation in the RET proto-oncogene. The male to female ratio is about equal.
MEN2 has three distinct subtypes, defined by the range of body tissues affected, with MEN2A being much the most common:
- MEN Type 2A (MEN2A)
- Familial Medullary Thyroid Cancer (FMTC) - MTC occurs alone
- MEN Type 2B (MEN2B)
Clinical Features
It was John H. Sipple, an American physician who first described this association between thyroid cancer and phaeochromocytoma in 1961, but it was not discovered until 1965 that the thyroid cancer involved was medullary thyroid carcinoma. Three years later the grouping of these two tumours with parathyroid hyperplasia was recognized as Multiple Endocrine Neoplasia Type 2 (MEN2).
The distinction between MEN2A (95% of cases) and MEN2B (5% of cases) however, was not made until 1975. The diagnosis of the two syndromes depends on the demonstration of the combination of characteristic tumours, plus other associated features in the case of MEN2B.
Some of the distinguishing differences are summarised below:
- Gastrointestinal, skeletal, and dermatological abnormalities only occur in MEN2B patients
- MEN2A patients have a less virulent form of medullary thyroid cancer (MTC) than MEN2B patients
- MEN2A patients may also have primary hyperparathyroidism, which is extremely uncommon in MEN2B patients
Biochemically, the tumours that arise in patients with these genetic syndromes are similar to those with the sporadic forms of the tumours, described on their own webpages. There are no distinct biochemical markers that allow identification of familial versus non-familial forms of the tumours.
1. Medullary thyroid cancer (MTC)
The characteristic tumour of MEN2 is MTC, which is present in all three subtypes. FMTC, which is a subset of MEN2A, has only medullary thyroid cancer without other endocrine tumours, and is distinguished from sporadic MTC by the strong family history.
Nearly all MEN2A patients develop medullary thyroid cancer. This is often the first expressed abnormality and usually occurs in the second or third decade of life. The medullary thyroid cancer in MEN2A patients is typically bilateral and multicentric, in contrast to sporadic medullary thyroid cancer which tends to be unilateral.
MTC may present as a lump in the neck, have obstructive symptoms like swallowing difficulty (dysphagia), or it can be entirely asymptomatic, being diagnosed unexpectedly after surgery or as part of a screening program. More details can be found on the MTC webpage by clicking the link above.
Diarrhoea occurs in 30% of patients, associated with elevated blood calcitonin or tumour-related secretion of hormones such as serotonin and prostaglandins. Patients may also present with unusual skin changes such as lichen amyloidosis (Fig. 1).
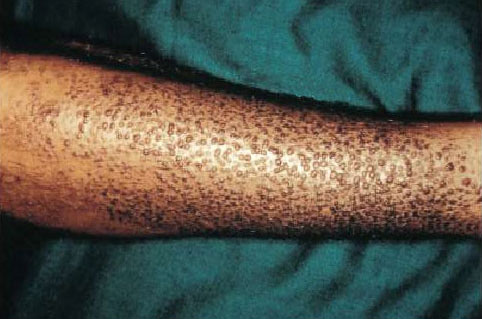 Fig.1: Lichen amyloidosis seen with MEN2A 2. Phaeochromocytoma
Fig.1: Lichen amyloidosis seen with MEN2A 2. Phaeochromocytoma
Phaeochromocytomas can occur in both MEN2A and MEN2B patients. They are present in approximately 50% of MEN2A patients. They are bilateral in 60-80% of patients, compared with 10% of patients with sporadic phaeochromocytomas (Fig. 2).
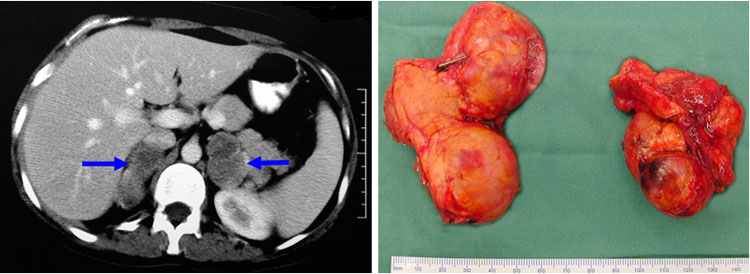 Fig.2: CT scan (left) and specimen (right) of bilateral phaeochromocytomas, typical of MEN2
Fig.2: CT scan (left) and specimen (right) of bilateral phaeochromocytomas, typical of MEN2
Phaeochromocytomas tend to be diagnosed at the same time as the medullary thyroid cancer or several years later (both primarily occurring in the second or third decade of life). The phaeochromocytomas of MEN2A patients are nearly all benign.
If phaeochromocytomas develop, an increase in blood pressure and heart rate may be the only signs, and these increases can be chronic or episodic. Some patients have episodes of sweating and headaches, or a 'feeling of impending doom'. More details can be found on the phaeochromocytoma webpages in the Adrenal section, by clicking the link above.
3. Hyperparathyroidism
Excessive parathyroid hormone secretion, causing hypercalcaemia (primary hyperparathyroidism) can occur in MEN2. Unlike sporadic primary hyperparathyroidism, where a single tumour (adenoma) in one gland is usually seen, the change in MEN2 is hyperplasia (excess growth) of all four glands.
This parathyroid hyperplasia occurs in about 15-30% of MEN2A patients, but is extremely rare in MEN2B. Parathyroid disease is less common in MEN2A than phaeochromocytoma.
In many patients, such hyperparathyroidism can be clinically silent, but on close questioning symptoms can often be elucidated. It is worth noting that more than half of these patients have no abnormality in their calcium levels and their parathyroid abnormalities are found during thyroidectomy for medullary thyroid cancer. The parathyroid hyperplasia found in MEN2A causes the same kind of primary hyperparathyroidism as in MEN1. More details about hyperparathyroidism can be found by following the link above.
4. Associated Features of MEN2B
- Marfanoid habitus: this is a characteristic body shape where the patient is tall and slight, with long limbs and digits, characteristically seen in a connective tissue disorder called Marfan's syndrome. Affected patients have a high-arched palate, pectus excavatum, bilateral pes cavus, and scoliosis, but it is not associated with the aortic aneurysms or dislocated lenses in the eye seen in true Marfan's syndrome
- Neuromas (small benign tumours of nervous tissue) on the eyelids, conjunctiva, nasal and laryngeal mucosa, tongue, and lips are frequent findings (Fig. 3) - they are treated conservatively
- Megacolon: this is a condition caused by a problem with the nerves supplying the large bowel, resulting in chronic distension and therefore chronic constipation
- Patients also have prominent hypertrophied lips leading to a characteristic facial appearance
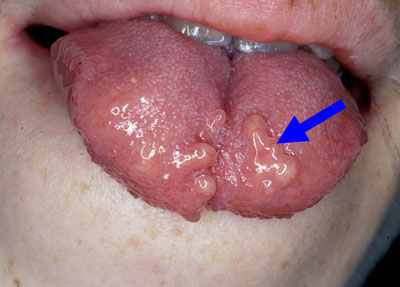 Fig.3: Tongue neuromas seen in MEN2B
Fig.3: Tongue neuromas seen in MEN2B
Diagnosis
Investigation of MEN2 syndrome is carried out to determine two things:
- to identify an individual at risk by genetic screening for RET mutations, and to determine whether other family members are at risk, or already affected (see Genetic Screening)
- which of the endocrine glands are affected by tumour by biochemical screening
For individuals identified with a mutation or for individuals who are at risk, biochemical screening consists of:
- calcitonin level to look for MTC
- plasma metanephrine concentrations to look for phaeochromocytoma
- serum calcium and parathyroid hormone (PTH) level to look for hyperparathyroidism
Once these tests are done further testing may be required to guide the appropriate treatment:
- if calcitonin is normal, screening for MTC is done with the pentagastrin or calcium stimulation test, which can unmask tumours only secreting small amounts of calcitonin (see MTC)
- if catecholamines are elevated, then imaging with CT and Dotate PET scans may be required (see investigation of phaeochromocytoma)
- suspected hyperparathyroidism may require sestamibi and ultrasound scanning (see hyperparathyroidism diagnosis)
Treatment
The treatment of MEN2 associated tumours folllows the standard treatment protocols that apply to each of the individual tumours.
Tumours of the parathyroid can be dealt with when the total thyroidectomy for the MTC is performed, but patients with phaeochromocytomas must be rendered safe by medication and adrenal tumours removed first, before total thyroidectomy can be carried out.
Following treatment, the patient and their affected relatives need careful and life-long follow up, with physical examination and routine biochemical testing:
- make sure all relatives have been appropriately screened for mutations in the RET gene
- patients diagnosed with MTC require serial calcitonin (with or without provocative stimulation testing) and carcinoembryonic antigen (CEA) levels to assess for persistent or recurrent disease - the 5- and 10-year survival rates in patients with MTC and MEN2A are approximately 90% and 75%, respectively
- annual plasma metanephrines to detect phaeochromocytoma at the earliest age possible
- annual testing of serum calcium and PTH levels from the age of 10 years - persistent or recurrent hyperparathyroidism is unusual and less likely to occur in MEN2A patients than in MEN1 patients